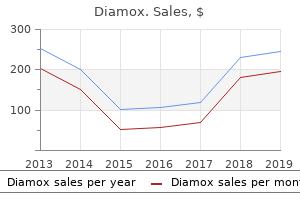
"Buy diamox 250 mg with visa, treatment 1st line".
By: X. Jens, M.B.A., M.D.
Vice Chair, Georgetown University School of Medicine
Functional limitation is outlined as a clinically appreciable limited vary of movement of a joint secondary to contracture or to skin and subcutaneous tissue involvement symptoms stroke discount 250mg diamox amex, but not as a result of medicine park oklahoma purchase diamox 250 mg with mastercard abnormality Table 57 medicine 8 - love shadow cheap 250 mg diamox free shipping. Early inflammatory part Granuloma annulare Early extragenital lichen sclerosus Lyme disease� erythema migrans Mycosis fungoides Cutaneous mastocytosis Radiation dermatitis Fixed drug eruption Sclerotic section Necrobiosis lipoidica Pretibial myxoedema Hyperpigmented section Postinflammatory hyperpigmentation Actinic lichen planus Caf� au lait macule Atrophic section Lipodystrophy Steroidinduced atrophy Lupus profundus Acrodermatitis chronica atrophicans Panniculitis (late stage) Clinical features fifty seven medicine vile buy diamox 250mg without prescription. In a 3rd of circumstances neurological adjustments have been unrelated to the site of skin involvement. These findings are just like those of a systematic evaluate of neurological involvement in 224 printed instances of linear morphoea in which seizures (42%) and headache (19%) predominated, however cranial nerve palsies, hemiparesis and neuropsychiatric involvement had been additionally documented [300,301]. Both ipsilateral and contralateral bone thinning, cerebral atrophy, white matter lesions, focal subcortical calcifications and meningocortical changes have been described [301]. Scalp and calvarial abnormalities similar to atrophy, T2 hyperintensities, calcifications and ipsilateral cerebral atrophy are probably the most commonly reported [154]. In one research of 16 sufferers these included malocclusion (94%), an overgrowth tendency of the anterior lower third of the face (82%), abnormal mastication (69%), dental anomalies (63%), skeletal asymmetry (56%), bone involvement (50%) and temporomandibular joint involvement (19%) [302]. Enophthalmos [277], choroidal and retinal folding, hyperopia, retinal vasculitis, glaucoma and third nerve palsies are additionally documented [276]. Nine of the children with Raynaud phenomenon had a couple of extracutaneous manifestation, including arthritis, gastro oesophageal reflux and cardiac arrhythmia. Symptomatic gastrooesophageal reflux, confirmed on appropriate investigations, was identified in 1. As with the Raynaud phenomenon, roughly 60% of those youngsters had other concomitant extracutaneous features. Four per cent of kids had a couple of extracutaneous manifestation and again this occurred predominantly in sufferers with linear illness. Together these findings recommend a more widespread inflammatory and/or autoimmune course of in some forms of morphoea, in addition to a necessity for more systematic multiorgan baseline investigations. Psychological morbidity was highest in patients with generalized morphoea and eosinophilic fasciitis, and in these with extra severe disease, larger levels of pain and fatigue and a higher impact of illness on day by day life and social support [309]. The second research, in 277 adults and youngsters, confirmed that reductions in quality of life correlated with useful impairment and signs of active disease similar to ache and pruritus, independently of illness subtype, age and sex [282]. Disease course and prognosis Early research indicated that the duration of illness exercise, though variable, was often 3�5 years [35], with plaques usually resolving sooner than other subtypes. However, mean disease period of childhoodonset morphoea could additionally be twice as lengthy as that for adultonset disease (13. In a retrospective study of 113 adults and 126 children referred over a 20year period to 2001, children with mixed forms of illness had been extra likely to run a extra protracted and complex course, and relapse was extra frequent in generalized, deep and combined types [22]. In a retrospective chart evaluate of fifty two paediatric patients with linear morphoea seen between 1990 and 2010, although illness stabilized after a mean length of 5. Reactivation of illness was frequent, even after seemingly effective courses of methotrexate and corticosteroids, such that 31% of patients reported lively disease after 10 years [23]. The most recently printed retrospective chart review contains 344 patients of whom 119 had a childhood onset of illness. Disease recurrence occurred in 27% of the paediatriconset group and 17% of the adultonset group. The linear limb variant was related to a better risk of recurrence, regardless of age at the onset of illness [310]. The longterm impression of paediatriconset morphea was additional highlighted in a group of 27 grownup sufferers (mean age 30. Overall, 81% had persistent symptoms of pain, itch or numbness; 89% of sufferers had persistent disease exercise, steady in 8/27, and with a remitting�relapsing course in 16/27. Seven of 27 (29%) sufferers described flares of exercise triggered by trauma, being pregnant or discount or discontinuation of systemic remedy. Fifteen of 20 sufferers with linear disease had everlasting sequelae together with lowered range of movement and deep atrophy. These research highlight the need for cautious counselling of sufferers and their households and continued vigilance. Concomitant morphoea, significantly plaque and nodular varieties, have been reported in up to 6. Investigations the analysis of morphoea is largely clinical, however a quantity of investigations may be helpful in guiding management (Table fifty seven. A biopsy may be undertaken if the prognosis, the depth of involvement or the degree of activity are in doubt. An incisional ellipse via the violaceous/inflammatory edge of a lesion, into the sclerotic centre and lengthening down into the subcutis, should be taken and the location clearly indicated for the pathologist.
Histologically medicine hat weather 250 mg diamox visa, amorphous masses that bind elastic tissue stains can be seen traversing the epidermis treatment yeast infection male buy diamox australia. Some of the lesions show a central despair containing an adherent necrotic plug medicinebg generic diamox 250 mg. It is most often seen with diabetes or earlier than medications ms treatment buy genuine diamox line, throughout or after dialysis for renal failure [7]. Eleven per cent of seventy two British sufferers on renal dialysis developed a perforating dermatosis [8]. Many individual reports of affiliation with malignancy, an infection or inflammatory circumstances have been published, however usually additionally with renal failure or diabetes [9]. This mainly affects kids, with the formation of 2�5mm papules, often on the limbs. Investigations Histology of acquired perforating dermatoses may show follicular and nonfollicular lesions with broad or narrow ulcer craters [9]. Degenerate collagen, elastic tissue and keratin are seen blended with an unidentified clear material, which has been regarded by some as an accumulation of a metabolite [2]. Changes diagnosed as perforating folliculitis, reactive perforating collagenosis or Kyrle illness (hyperkeratosis follicularis et parafollicularis in cutem penetrans) could all be discovered [11�13]. Clinical features Cases may be sporadic [3] or familial with probable autosomal dominant inheritance [2,7,8], early or late onset, transient or persistent. Other agents reported to be effective embody allopurinol [17] and doxycycline [18]. Hyperkeratotic spicules on the face, significantly on the nostril, are follicular and sometimes related to paraproteinaemia, multiple myeloma and croyglobulinaemia, however may be also idiopathic [10,11]. A familial type of filiform keratosis has been described to be associated with thickened nails, plantar hyperkeratosis, joint laxity and lengthy fingers [16]. Investigations Histologically, there are focal areas of compact orthohyperkeratotic spicules largely arising from a pointed epidermal elevation. Parakeratosis may be present [16,20] however the utilization of the term porokeratosis is deceptive [5,6]. Hyperkeratotic spicules related to paraproteinaemia reveal eosinophilic inclusions in the hyperkeratotic columns that symbolize immunoglobulin deposits [10]. Flegel disease Definition and nomenclature Flegel illness is a uncommon and benign cornification disorder of older individuals characterized by a quantity of reddishbrown keratotic papules affecting the extremities [1]. In the periphery of the lesions, the dermis is acanthotic with collarettelike elongated rete ridges. Upon electron microscopy membrane coated granules (Odland bodies) appear reduced and malformed at least in evolving lesions [1,2,3,5,6]. Despite the strong genetic element within the disorder, no reports identifying a candidate gene have appeared to date. A low proliferation price of keratinocytes along with downregulation of filaggrin, loricrin and highmolecularweight keratins and loss of the keratin sample in the sexy layer suggests a retention hyperkeratosis and complicated dysregulation of the epidermal differentiation [3]. Circumscribed palmoplantar hypokeratosis Pathophysiology this condition is considered a localized keratinization dysfunction of an expanding clone of keratinocytes [1]. Acanthosis, dilated tortuous capillaries and coarse keratohyalin granules are additionally suggestive of a viral origin. The lesions might unfold to the higher a half of the legs and thighs, additionally disseminating over the arms and trunk or concha of the ear. Differential prognosis consists of the leukodermic macules in Darier illness in dark pores and skin [7]. Larger papules with a mosaic pattern and acral distribution have been diagnosed as mosaic acral keratosis (see additionally Focal acral hyperkeratosis earlier) [8]. Investigations Histological findings were marked orthokeratotic hyperkeratosis, tenting and papillomatosis of the dermis, and mild acanthosis. Schedel, Department of Dermatology, University Hospital M�nster, M�nster, Germany. Differential prognosis the most important differential analysis is Paget disease. Investigations Histologically, an abrupt thinning of the stratum corneum over a diminished granular layer types a sharp stair between normal and involved skin [6]. Malignant transformation is unlikely although susceptibility to photocarcinogenesis has been assumed [7,8].
Less severely affected kind A sufferers have juvenile or adultonset neurological illness symptoms meaning buy discount diamox. Type B patients current as children or adults with splenomegaly or hepatosplenomegaly; complications embody interstitial lung illness ombrello glass treatment purchase diamox on line, poor development medications not to take with blood pressure meds purchase diamox 250mg with visa, hyperlipidaemia and thrombocytopenia symptoms 7 days after embryo transfer purchase diamox discount. The pores and skin could also be involved in sorts A or B, with patches of waxy induration and brownish yellow pigmentation. Severe cases present as neonates with hydrops fetalis or hypotonia, hepatosplenomegaly and facial dysmorphism (including macroglossia, gum hypertrophy and a depressed nasal bridge). The mildest instances present in late childhood with dysarthria, dystonia and skeletal dysplasia (affecting the backbone and hip). Telangiectasia and intensive Mongolian blue spots have also been reported in infantile circumstances. Patients normally present in early infancy with a hoarse cry, painful swollen joints and subcutaneous nodules. The mostly affected joints are those of the hand and wrist, elbows, knees and ankles. The subcutaneous nodules could also be associated with erythematous papules and are generally close to affected joints and over pressure factors, such because the occiput and decrease backbone. Histology reveals granulomas containing massive, foamy histiocytes; electron microscopy exhibits that these have cytoplasmic vacuoles containing curvilinear inclusions (Farber bodies) [17]. Most patients have psychomotor retardation, poor weight acquire and die in early childhood from respiratory infections. Introduction and basic description this could be a uncommon Xlinked lysosomal storage dysfunction, characterised by angiokeratomas and multisystem problems [19]. Affected males normally current in childhood with episodes of pain in the extremities, adopted by the looks of angiokeratomas. Many heterozygous females also develop signs, although the onset is often later. Pathophysiology A deficiency of galactosidase A prevents the degradation of glycosphingolipids with terminal galactose residues, predominantly globotriaosylceramide (Gb3). Gb3 accumulation in vascular endothelial, perithelial and smooth muscle cells results in aneurysmal dilatation of blood vessels, ischaemia and infarction. Glycosphingolipids additionally accumulate within the renal glomeruli and tubules, cardiac muscle, autonomic ganglion cells and corneal epithelium. Investigations the diagnosis is established by enzyme assays on leukocytes (or cultured cells). Niemann�Pick cells are typical examples � large, usually mononucleate histiocytes, whose cytoplasm is crammed with lipid droplets; they stain readily with Sudan stains and include doubly refractile materials. Ultrastructurally, the cytoplasm of Niemann� Pick cells accommodates granular lipid inclusions which will seem lamellar [15]. Ultrastructurally, Gaucher cells have vesicles that comprise twisted tubular constructions [18]. Clinical options the first symptoms are normally episodes of extreme burning ache in the palms and soles (acroparesthesiae). These occur in 70�85% of male patients, usually beginning at between 5 and 15 years of age, although diagnosis is commonly delayed [21]. Acroparesthesiae occur in 50�70% of feminine patients, with a imply age of onset of 15 years [22]. Painful crises are sometimes triggered by fever or exertion and should last hours or days. Angiokeratoma corporis diffusum happens in 65�70% of male patients and 35�40% of feminine sufferers [21,22]. Lesions may also be discovered on the higher arms, around the border of the lips, across the nail folds and on the palms and soles � these are usually macular angiomas with minimal hyperkeratosis. In girls, lesions are most frequent on the trunk and proximal limbs; genital lesions are uncommon. Telangiectases are present in 23% of male sufferers and 9% of female patients, usually on the lips, buccal mucosa, ears or conjunctiva [23]. Anhidrosis or hypohidrosis happens in 53% of male sufferers and 28% of female patients, often beginning in the third decade [23]. Miglustat is an oral drug that decreases the accumulation of glucocerebroside by lowering the synthesis of glycosphingolipids (substrate discount therapy) [12].
Failure to respond adequately to local anaesthetic is reported more commonly on this subgroup [48] medications like abilify purchase diamox 250mg on line. Detection of abnormal pyridinoline crosslinks in urine can be used as a diagnostic help [62] treatment uterine cancer 250 mg diamox visa.
[newline]Clinical features include gentle velvety hyperextensible pores and skin and elevated joint mobility symptoms quivering lips buy generic diamox from india. Eye manifestations embrace microcornea treatment anemia quality diamox 250 mg, glaucoma, keratoconus and ocular fragility. Bleeding might occur from major wounds, and there could additionally be delayed motor growth [63]. Other features include prematurity (due to rupture of friable placental membranes), easy bruising (which could result in the mistaken accusation of kid abuse) [49] inguinal hernia and pneumothorax [50,51]. The major complications come up following spontaneous rupture of large arteries, colon and gravid uterus. Most deaths follow arterial dissection or rupture, primarily of the thoracic and belly vessels. In the identical series, problems of being pregnant led to death within the peripartum period in 12 of eighty one girls who had a complete of 183 pregnancies [24]. Overlap happens with adducted thumb�clubfoot syndrome and more than likely represents a continuous phenotypic spectrum. Ehlers�Danlos syndrome spondylocheiro dysplastic form Patients exhibit thin skin with easy bruising and wrinkling of the palms, small joint hypermobility typically with development to contractures, tapering fingers, platyspondyly and widened metaphyses [68]. There is significant skin redundancy, leading to one of many key features of arthrochalasia kind namely a crisscross sample on the palms and soles [70]. The biochemical defect is unknown [54], and lysyl oxidase has been proven to be decreased in one [55] however not different families [56]. Until a precise genetic or biochemical abnormality is discovered, the standing of this type should remain in question [23]. This autosomal recessive situation was the first true disorder of collagen construction to be described [56�58]. Facial appearance is characterised by blue sclera, puffy eyelids, epicanthic folds, downslanting palpebral fissures, micrognathia with gingival hyperplasia and dental anomalies [76]. Other features embody umbilical hernia, variable joint laxity that turns into extra outstanding with age, delayed closure of the fontanelles, brief stature and brachydactyly. Two cases with congenital skull fractures and pores and skin lacerations after start have been reported [77,78]. Electron microscopy of the pores and skin confirms decreased collagen content, with abnormal variation in collagen fibril diameter and shape [82]. It is inherited in an autosomal dominant style and has been mapped to chromosome 12p13 in three out of 5 households, indicating genetic heterogeneity [83]. Fibronectindeficient kind (Ehlers�Danlos syndrome X) Only one family has been identified to date with this autosomal recessive dysfunction. Skin and joint modifications are mild, however bruising happens readily due to faulty platelet aggregation. Some patients have composite fibrils in the dermal collagen, probably because of a defect in fibronectin interactions [84]. Patients lack the total phenotype of progeria of the extra, welldefined progeroid syndromes. Patients have hyperextensible pores and skin, bruising and joint laxity, however no scarring [94,95]. A multidisciplinary strategy is required with involvement of acceptable specialists. Differential diagnosis Ehlers�Danlos syndrome should be distinguished from cutis laxa (see Chapter 79), by which the skin hangs in flaccid redundant folds. Hyperelastic pores and skin is a feature of Turner syndrome but the dwarfism, cubitus valgus and webbed neck are distinctive. Hyperelastic pores and skin with abnormal elastic fibres in the papillary dermis has been reported within the uncommon cartilage�hair hypoplasia syndrome [99].
Multiple medicine 4839 order diamox 250 mg overnight delivery, ovalshaped osteolytic lesions with welldefined margins in the metaphysis and/or diaphysis of the long tubular and flat bones are sometimes seen [127�129] medications emt can administer order cheap diamox. They manifest as multilobulated medications with aspirin purchase diamox 250mg with amex, welldefined lesions which are translucent medications used to treat depression buy diamox with mastercard, soft, however only slightly compressible on palpation. Capillarolymphatic malformation or angiokeratoma circumscriptum is a hyperkeratotic plaque usually positioned on the extremity. It can abruptly turn into painful and enlarge as a outcome of intralesional bleeding or infection. It manifests as muscular atrophy, bone pain, fractures, pleural effusion, ascites and cerebrospinal fluid rhinorrhoea. Management Bacterial an infection must be managed with systemic antibiotics and ache medicines. Complete surgical excision may be proposed depending on the anatomical site and extension of the lesion. Blood could be seen within the areas after intracystic bleeding or in a combined lymphaticovenous malformation. The unravelling of the underlying aetiopathogenic mechanisms permits the design of focused molecular therapies. Epidemiology the prevalence is unknown, however this is probably probably the most frequent type of major lymphoedema [130]. A few households with pleural effusion, chylous ascites or hydrops have been reported [153�155]. Other associated features include hydrocele, outstanding veins, upslanting toenails and papillomatosis [156]. Investigations Lymphoscintigraphy has replaced lymphography in the investigation of lymphoedema. Management the associated vascular anomalies are managed equally to the isolated counterparts. The onset of lymphoedema is in childhood or adolescence, and later in males than in females [159]. Other associated options are ptosis, cellulitis and venous insufficiency; recurrent skin infections are widespread. Cellulitis and recurrent cellulitis are handled with appropriate antibiotics and prophylactic antibiotics, respectively. Suction assisted lipectomy in combination with postoperative elastic compression can be helpful [164]. Understanding the molecular mechanisms at the origin of the completely different subtypes of main lymphoedema is a vital step in creating targeted therapies sooner or later. When symptomatic, distichiasis can cause corneal irritation, an infection and photophobia. Other related options are varicose veins, cardiac malformations, cleft palate, ptosis and yellow nails [158,165]. Introduction and common description this syndrome is an autosomal recessive, generalized, lymphatic dysplasia characterised by intestinal lymphangiectasias associated with extreme and progressive lymphoedema of the limbs, genitalia and face. Mutations have been recognized in a subset of patients solely, suggesting genetic heterogeneity. Clinical features In addition to generalized lymphatic dysfunction, distinct facial features � similar to flat face, low nasal bridge, hypertelorism, epicanthal folds, small mouth, and dental and ear anomalies � are noticed. Syndactyly, camptodactyly, craniosynostosis, hypothyroidism and ectopic kidney may be current. Clinical features the dysfunction is characterized by lymphoedema in the decrease extremities, which develops throughout childhood or at puberty, sparse hair, absent eyelashes and eyebrows, and telangiectasias within the palms, soles, scalp and scrotum [168]. Proteinlosing enteropathy secondary to intestinal lymphangiectasias is handled with cautious dietary management, including fats restriction, mediumchain triglyceride supplementation or parenteral diet [176]. It is an autosomal dominant dysfunction with incomplete penetrance and intrafamilial variable expressivity. Epidemiology Primary lymphoedema with myelodysplasia is a really rare syndrome with just a few families reported.
Purchase diamox 250 mg without a prescription. Questionnaire Formation for Data Collection & Statistical Analysis (For Beginners in Urdu).