Medical Instructor, Creighton University School of Medicine
Chapter forty three � Drugs Used for Psychopharmacologic Therapy 825 Fluvoxamine Fluvoxamine is efficient in the management of obsessivecompulsive issues. Bupropion Bupropion, which is structurally associated to amphetamine, is effective within the remedy of major despair, producing enchancment in 2 to four weeks. The mechanism of motion of bupropion is obscure but could embrace inhibition of dopamine and norepinephrine reuptake. At excessive doses, a modest however persistent enhance in diastolic blood stress occurs in 5% to 7% of sufferers. Some research have advised that venlafaxine could also be helpful in patients with neuropathic pain. The elimination half-time is 5 hours and that of its energetic metabolite is eleven hours. Duloxetine Duloxetine is a serotonin and noradrenaline reuptake inhibitor, just like venlafaxine. Trazodone Trazodone inhibits serotonin reuptake and may also act as a serotonin agonist via an energetic metabolite. Common side effects of trazodone embrace sedation, orthostatic hypotension, nausea, and vomiting. This drug lacks effects on conduction of cardiac impulses however on uncommon occasions has been related to cardiac dysrhythmias. Nefazodone Nefazodone is chemically associated to trazodone however with fewer a1-adrenergic blocking properties. The danger of sedation and priapism may be lower than in sufferers treated with trazodone. Measurement of plasma drug levels for the tricyclics imipramine, desipramine, and nortriptyline could be helpful in guiding therapeutic choices. It is preferable to taper tricyclic and tetracyclic antidepressants during a 4-week period to keep away from the chance of withdrawal signs (chills, coryza, muscle aches). These signs have been attributed to supersensitivity of the cholinergic nervous system. Chronic Pain Syndromes the tricyclic antidepressants (especially amitriptyline and imipramine), in doses lower than these used to deal with despair, may be useful within the remedy of chronic neuropathic pain and different continual pain syndromes including fibromyalgia. Structure�Activity Relationships the construction of tricyclic antidepressants resembles that of local anesthetics and phenothiazines. Similar to native anesthetics, tricyclic antidepressants embody a hydrophobic portion linked to an amide via a linear intermediate moiety. Tricyclic denotes the three-ring chemical structure of the central portion of the molecule. Imipramine, which is the prototype of the tricyclic antidepressants, differs from phenothiazine only within the replacement of the sulfur atom with an ethylene linkage to produce a seven-membered central ring. Maprotiline is a tetracyclic antidepressant with a scientific profile that resembles imipramine. Mechanism of Action Tricyclic antidepressants act at several transporters and receptors, but their antidepressant impact is likely produced by blocking the reuptake (uptake) of serotonin and/or norepinephrine at presynaptic terminals, thereby growing the provision of these neurotransmitters. These medication could be categorized into tertiary amines, which inhibit reuptake of each serotonin and norepinephrine (amitriptyline, imipramine, clomipramine) and secondary amines, which are primarily norepinephrine reuptake inhibitors (desipramine, nortriptyline). Despite the immediate onset of this impact, the development of a therapeutic antidepressant effect is inexplicably delayed for 2 t o 3 w eeks. Furthermore, some medicine without results on uptake of biogenic amines are effective antidepressants. It appears doubtless that potentiation of monoaminergic neurotransmission in the mind is just an early event in a complex cascade of events that finally results in an antidepressant impact. Indeed, chronic administration of these medicine is related to (a) decreased sensitivity of postsynaptic b1 and serotonin2 receptors and of presynaptic a2 receptors, and (b) increased sensitivity of postsynaptic a1 receptors. Pharmacokinetics Tricyclic antidepressants are efficiently absorbed from the gastrointestinal tract after oral administration, reflecting high lipid solubility. Therapeutic plasma concentrations (parent drug plus the pharmacologically active demethylated metabolites) are a hundred t o 300 ng/ mL, whereas toxicity is likely at ranges greater than 500 ng/mL. Tricyclic antidepressants are strongly certain to plasma and tissue proteins, which, together with high lipid solubility, leads to a large volume of distribution (up to 50 L/kg) for these medicine. The lengthy elimination half-time (17 to 30 hours) and wide range of therapeutic plasma concentrations make once-daily dosing intervals eff ctive. Metabolism Tricyclic antidepressants are oxidized by microsomal enzymes within the liver with subsequent conjugation with glucuronic acid. The individual variation in rate of metabolism Chapter forty three � Drugs Used for Psychopharmacologic Therapy 827 between patients is 10- to 30-fold. The elimination of tricyclic antidepressants occurs over a number of days, with 1 week or longer required for excretion. Both these lively compounds are inactivated by oxidation of hydroxy metabolites and by conjugation with glucuronic acid. Nortriptyline, which is the pharmacologically active demethylated metabolite of imipramine and amitriptyline, can accumulate to ranges that exceed the precursors. Doxepin also seems to be transformed to an energetic metabolite, nordoxepin, by demethylation. Individual variation in the incidence and sort of side effects could additionally be associated to the plasma concentrations of the tricyclic antidepressant and its energetic metabolites. Anticholinergic Effects the anticholinergic results of tricyclic antidepressants are prominent, especially at excessive doses. Amitriptyline causes the very best incidence of anticholinergic effects (dry mouth, blurred vision, tachycardia, urinary retention, slowed gastric emptying, ileus), whereas desipramine produces the fewest such results (see Table 43-2). Anticholinergic delirium could occur in elderly sufferers even at therapeutic doses of those medicine. Serious anticholinergic toxicity could replicate the outcomes of polypharmacy with a couple of anticholinergic drug (over-the-counter preparations to treat diarrhea or insomnia). Elderly patients have larger sensitivity to anticholinergic and different receptor effects in contrast with younger patients being handled with tricyclic antidepressants. Cardiovascular Effects Orthostatic hypotension and modest will increase in heart rate are the most typical cardiovascular unwanted aspect effects of tricyclic antidepressants, presumably refl cting druginduced inhibition of norepinephrine reuptake into presynaptic nerve terminals. The danger of hypotension during general anesthesia in sufferers treated with tricyclic antidepressants is low however has been reported. Direct cardiac depressant effects might replicate quinidinelike actions of tricyclic antidepressants on the heart. Conceivably, there is also enhancement of depressant cardiac results of anesthetics by tricyclic antidepressants. Quinidine-like properties of tricyclic antidepressants are thought to replicate slowing of sodium ion flux into cells, leading to altered repolarization and conduction of cardiac impulses. Central Nervous System Effects Sedation associated with tricyclic antidepressant remedy could additionally be desirable for management of depressed sufferers with insomnia. Amitriptyline and doxepin produce the best degree of sedation (see Table 43-2). Tricyclic antidepressants, particularly maprotiline and clomipramine, decrease the seizure threshold, elevating the question of the advisability of administering these medication to sufferers with seizure problems or to these receiving medicine that may produce seizures. Children seem to be especially susceptible to the seizure-inducing results of tricyclic antidepressants. Extrapyramidal reactions are uncommon, although a fine tremor develops in about 10% of sufferers, especially the aged. Drug Interactions the anticholinergic results and catecholamine uptake blocking properties of tricyclic antidepressants are most likely to be answerable for drug interactions. Drug interactions may be prominent with (a) sympathomimetics, (b) inhaled anesthetics, (c) a nticholinergics, (d) a ntihypertensives, and (e) opioids. Binding of tricyclic antidepressants to plasma albumin could be decreased by competitors from different medication, together with phenytoin, aspirin, and scopolamine. Sympathomimetics the systemic blood stress response to the administration of sympathomimetics to patients treated with tricyclic antidepressants is advanced and unpredictable. Although acute administration of tricyclic antidepressants will increase sympathetic nervous system synaptic exercise as a result of norepinephrine reuptake blockade, persistent administration of these medicine might result in decreased sympathetic nervous system transmission due to downregulation of b-adrenergic receptors. Smaller than usual doses of direct-acting sympathomimetics which would possibly be titrated to a particular hemodynamic response are recommended. Conversely, standard sympathomimetics may not be efficient in restoring systemic blood stress in sufferers chronically handled with tricyclic antidepressants as a outcome of adrenergic receptors are either desensitized or catecholamine shops are depleted. In these patients, a potent direct-acting sympathomimetic corresponding to norepinephrine may be the solely effective management for hypotension. Likewise, the dose of exogenous epinephrine needed to produce cardiac dysrhythmias throughout anesthesia with a unstable anesthetic is decreased by tricyclic antidepressants.
Calcium Channel Blockers Calcium channel blockers (also known as calcium entry blockers and calcium antagonists) are a various group of structurally unrelated compounds that selectively interfere with inward calcium ion motion throughout myocardial and vascular easy muscle cells. Commercially available calcium channel blockers are classified based mostly on chemical construction as phenylalkylamines, dihydropyridines, and benzothiazepines (Table 19-5 a nd. The a1 subunit varieties the central a half of the channel and offers the principle pathway for calcium ion entry into cells. All clinically used calcium channel blockers bind to a novel website on the a1 subunit of the L-type calcium channels and thus diminish entry of calcium ions into cells. These structurally diverse medication differ of their tissue selectivity, their binding web site location on the a1 subunit, and their mechanism of calcium blockade. Voltage-gated calcium ion channels are current in the cell membranes of skeletal muscle, vascular clean muscle, cardiac muscle, mesenteric muscle, glandular cells, and neurons. Thus, blockade of gradual calcium channels by calcium channel blockers predictably results in slowing of the heart fee, reduction in myocardial contractility, decreased pace benzothiazepines are selective for the atrioventricular node, whereas the dihydropyridines are selective for the arteriolar beds. The varied calcium channel blockers differ when it comes to unwanted effects, traditional doses, metabolism, and period of action (Tables 19-6 and 19-7). Chapter 19 � Sympatholytics 491 Table 19-6 Comparative Pharmacologic Effects of Calcium Channel Blockers Verapamil Systemic blood stress Heart charges Myocardial depression Sinoatrial node melancholy Atrioventricular node conduction Coronary artery dilation Peripheral artery dilation Decrease Decrease Moderate Moderate Marked Moderate Moderate Nifedipine Decrease Increase to no change Moderate None None Marked Marked Nicardipine Decrease Increase to no change Slight None None Greatest Marked Diltiazem Decrease Decrease Moderate Slight Moderate depression Moderate Moderate of conduction of cardiac impulses through the atrioventricular node, and vascular easy muscle relaxation. The intracellular calcium combines with calmodulin, the calcium-binding protein, to kind the calcium-calmodulin advanced. This complex activates myosin and causes the formation of cross-bridges with actin. Pharmacologic Effects the pharmacologic results of calcium channel blockers could also be predicted by contemplating the traditional position of calcium ions in the production of motion potentials, particularly in cardiac cells. It is predictable that calcium channel blockers will produce decreased myocardial contractility, decreased coronary heart price, decreased activity of the sinoatrial node, decreased fee of conduction of cardiac impulses via the atrioventricular node, and vascular clean muscle relaxation with related vasodilation and reduces in systemic blood pressure. All of the calcium channel blockers are efficient for the remedy of coronary artery spasm. Calcium channel blockers lower vascular smooth muscle contractility, thereby rising coronary blood move and inflicting peripheral vasodilation with reductions in systemic vascular resistance and systemic blood pressure. These druginduced responses contribute to the antiischemic results characteristic of calcium channel blockers. Calcium channel blockers are additionally efficient for the treatment of continual steady angina pectoris caused by fi ed obstructive coronary artery lesions and for the therapy of unstable angina pectoris. All calcium channel blockers exert adverse inotropic results, that are most vital with verapamil and diltiazem. Phenylalkylamines the phenylalkylamines bind to the intracellular portion of the L-type channel a1 subunit when the channel is in an open state and conceptually occlude the channel. The negative inotropic effects of verapamil appear to be exaggerated in sufferers with preexisting left ventricular dysfunction. Isoproterenol may be helpful to increase heart fee within the presence of drug-induced coronary heart block. Verapamil may precipitate ventricular dysrhythmias in sufferers with Wolff- arkinson-White syndrome. Clinical Uses Verapamil is efficient within the therapy of supraventricular tachydysrhythmias, refl cting its main site of motion on the atrioventricular node (see Chapter 21). Indeed, calcium channel blockers are as effective as b blockers in relieving angina pectoris. Verapamil is effective in the remedy of symptomatic hypertrophic cardiomyopathy with or with out left ventricular outfl w obstruction. Verapamil might decrease uterine blood flow, suggesting warning in the administration of this drug to parturients with impaired uteroplacental perfusion. Demethylated metabolites of verapamil predominate, with norverapamil possessing enough activity to contribute to the antidysrhythmic properties of the father or mother drug. In view of the almost complete hepatic metabolism of verapamil, almost not considered one of the drug seems unchanged within the urine. Conversely, an estimated 70% of an injected dose of verapamil is recovered in urine as metabolites and about 15% is excreted via bile. Chronic oral administration of verapamil or the presence of renal dysfunction leads to the buildup of norverapamil. The elimination half-time of verapamil is 6 to 12 hours, and this can be prolonged in sufferers with liver disease. In this regard, chronic remedy with verapamil has hardly ever been related to increased plasma concentrations of transaminase enzymes. Like nifedipine, verapamil is highly protein certain (90%), and the presence of other medicine (lidocaine, diazepam, propranolol) can increase the pharmacologically lively, unbound portion of the drug. As with other peripheral vasodilators, a reflex tachycardia attributed to sympathetic nervous system exercise or baroreceptor reflexes may be noticed with the acute administration of dihydropyridines. Nifedipine Nifedipine is a dihydropyridine spinoff with greater coronary and peripheral arterial vasodilator properties than verapamil. Unlike verapamil, nifedipine has little or no direct depressant impact on sinoatrial or atrioventricular node exercise. Peripheral vasodilation and the resulting lower in systemic blood pressure produced by nifedipine activate baroreceptors, resulting in elevated peripheral sympathetic nervous system exercise most often manifesting as an elevated heart fee. This elevated sympathetic nervous system activity counters the direct adverse inotropic, chronotropic, and dromotropic results of nifedipine. Nevertheless, nifedipine could produce excessive myocardial depression, particularly in sufferers with preexisting left ventricular dysfunction or concomitant remedy with a b-adrenergic antagonist drug. The presence of aortic stenosis can also exaggerate the cardiac depressant results of nifedipine. Clinical Uses Nifedipine is administered orally with a 10- to 30-mg dose producing an impact in about 20 minutes with a peak effect between 60 and ninety minutes. Nifedipine is used to treat patients with angina pectoris, especially that as a outcome of coronary artery vasospasm. Pharmacokinetics Absorption of an oral or sublingual dose of nifedipine is about 90%, with onset of an effect being detectable within about 20 minutes after administration (see Table 19-7). It is likely that a lot of the absorption of sublingual nifedipine is through the gastrointestinal tract from swallowed saliva. Hepatic metabolism is nearly full, with elimination of inactive metabolites principally in urine (about 80%) and, to a lesser extent, in bile. Side Effects the unwanted aspect effects of nifedipine embrace flushing, vertigo, and headache.
[newline]Less common unwanted aspect effects embrace peripheral edema (venodilation), hypotension, paresthesias, and skeletal muscle weak spot. Abrupt discontinuation of nifedipine has been related to coronary artery vasospasm. Nicardipine Nicardipine lacks effects on the sinoatrial node and atrioventricular node and has minimal myocardial depressant results. This drug has the greatest vasodilating effects of all of the calcium entry blockers, with vasodilation being particularly prominent in the coronary arteries. Combination Dihydropyridines the dihydropyridines prevent calcium entry into the vascular smooth cells by extracellular allosteric modulation of the L-type voltage-gated calcium ion channels. Of all the antianginal medication, the dihydropyridine calcium channel blockers produce the best dilatation of the peripheral arterioles. A long elimination half-time is the basis for the recommendation that about 72 hours should elapse before rising the oral dose. When nicardipine is administered as a tocolytic, it binds to the within of myometrial L-type voltage-dependent calcium ion channels, inflicting them to remain closed, and thus inhibits uterine contractility. Pulmonary edema related to salbutamol used as a tocolytic has also been reported in a parturient treated with nicardipine. Clinical Uses the lipid solubility of nimodipine and its capability to cross the blood�brain barrier is liable for the potential value of this drug in treating sufferers with subarachnoid hemorrhage. Cerebral Vasospasm the vasodilating impact of nimodipine on cerebral arteries is uniquely useful in preventing or attenuating cerebral vasospasm that usually accompanies subarachnoid hemorrhage. Symptoms of excessive nimodipine impact could be anticipated to be related to cardiovascular effects similar to peripheral vasodilation with associated systemic hypotension. Theoretically, druginduced cerebral vasodilation could evoke increases in intracranial strain, particularly in sufferers with preexisting decreases in intracranial compliance. Cerebral Protection Nimodipine has also been evaluated for cerebral protection after world ischemia as related to cardiac arrest. The theoretical foundation for contemplating calcium channel blockers for this objective is the statement that lack of oxygen interferes with maintenance of the traditional calcium ion gradient throughout cell membranes, main to an enormous increase (at least 200-fold) in the intraneuronal concentrations of this ion. In this regard, nimodipine is associated with improved neurologic outcome when administered to primates inside 5 minutes after experiencing 17 minutes of cerebral ischemia.
Ten years of hemovigilance reviews of transfusion-related acute lung injury within the United Kingdom and the impact of preferential use of male donor plasma. High-potency antihaemophilic factor concentrate prepared from cryoglobulin precipitate. A more potent human antihemophilic globulin concentrate: preparation and medical trial. Platelet transfusions for patients with haematological malignancies: who wants them Prophylactic platelet transfusion for prevention of bleeding in sufferers with haematological issues after chemotherapy and stem cell transplantation. The threshold for prophylactic platelet transfusions in adults with acute myeloid leukemia. Fresh-frozen plasma, cryoprecipitate, and platelets administration apply pointers improvement task drive of the college of yank pathologists. Important inflammatory mediators and interactions with endothelial cells orchestrate these events by upregulating adhesion molecules after hypoxic problem and manufacturing of inflammatory cytokines or pathogen metabolites that facilitate margination of leukocytes. Cytokines liberated as part of the inflammatory response induce modifications in integrins and enhance adhesion to the vascular wall adopted by transmigration across the vessel wall. Chemotactic factors launched domestically further attract neutrophils to areas of main tissue damage to kill invading organisms and remove necrotic tissue. Summary Blood and blood products are used extensively within the perioperative setting in surgical and trauma patients. Blood products and transfusions must be thought-about in the same method that we think about use of different drug therapies by carefully weighing their risks and benefits. The advanced setting in which we transfuse patients, the big selection of reasons to transfuse, and the different indications for blood product administration all additional emphasize the significance of contemplating risk and profit for each distinctive affected person. A multivariable mannequin for predicting the necessity for blood transfusion in patients present process first-time elective coronary bypass graft surgery. Red cell transfusion is associated with an increased threat for postoperative atrial fibrillation. Factors affecting posttransfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Animal model and scientific proof indicating low thrombogenic potential of fibrinogen focus (Haemocomplettan P). Impact of blood transfusions on inflammatory mediator release in sufferers undergoing cardiac surgery. A perspective on transfusion-related acute lung injury two years after the Canadian Consensus Conference. Evidence-based purple cell transfusion within the critically ill: high quality enchancment using computerized doctor order entry. Effective discount of transfusion-related acute lung damage risk with male-predominant plasma strategy within the American Red Cross (2006-2008). Levy Bleeding in a perioperative setting, following trauma or surgery, can arise from quite a few causes that embody activation of the coagulation, fibrinolytic, and inflammatory pathways; dilutional modifications; hypothermia; and surgical factors. Hemostatic operate and coagulation are complex and sometimes altered in by a quantity of occasions that occur within the perioperative setting. This chapter will concentrate on the position of procoagulants used in a perioperative setting. These agents competitively inhibit activation of plasminogen to plasmin, an enzyme that degrades fibrin clots, fibrinogen, and other plasma proteins. Most research reporting using antifibrinolytic agents are in cardiac surgical sufferers, but use in other patients, together with orthopedic patients have additionally been reported. Compared with placebo or no treatment, all three medication had been efficient in lowering the need for purple blood cell transfusion. The incidence of postoperative convulsive seizures at one institution was reported to increase from 1. Antifibrinolytic brokers even have been studied in other procedures, together with orthopedic surgery, and all three brokers scale back blood loss. Although a lot of the reported research included small numbers of sufferers and lacked Chapter 29 � Procoagulants 641 sufficient energy, larger meta-analysis and more modern data suggest that these agents characterize an necessary adjunct for decreasing bleeding and the need for allogeneic transfusions. A current meta-analysis examined the use of intravenous antifibrinolytics compared with placebo on pink blood cell transfusion requirement in orthopedic surgical procedure and the safety of these brokers, including venous thromboembolic danger. The main end result was in-hospital demise within 4 weeks of damage and was described as bleeding, vascular occlusion (myocardial infarction, stroke, and pulmonary embolism), multiorgan failure, head damage, or different causes. The recommended dose for girls with normal renal perform is two 650-mg tablets taken three times daily (3,900 mg per day) for a most of 5 days throughout monthly menstruation. In cardiac surgery, multiple randomized, placebo-controlled trials reported aprotinin as effective in lowering bleeding and allogeneic transfusions. As a result of this evaluation, the manufacturer, Bayer Inc, can resume the advertising of aprotinin in Canada. This impact, if Antifibrinolytic Agents: Aprotinin Aprotinin, a polypeptide serine protease inhibitor, inhibits plasmin and other serine proteases and has had a long historical past of use in different scientific purposes. Protamine Protamine is a polypeptide containing roughly 70% arginine residues and the one obtainable agent to reverse unfractionated heparin. This primary protein inactivates the acidic heparin molecule via a simple acid�base interplay. Protamine doses of 5 to 15 mg right now could also be efficient at reversing heparin rebound quite than the dose of 50 mg generally administered. Protamine can cause opposed reactions including anaphylaxis, acute pulmonary vasoconstriction and proper ventricular failure, and hypotension. For clot formation, thrombin cleaves the fibrinogen molecule, producing a soluble fibrin monomer which polymerizes to type a free network in trapping pink blood cells and a clot begins to kind. These platelets then turn out to be enmeshed throughout the fibrin strands, stabilizing the growing clot, and create the flexibility to cross-link and expand the clot and seal the bleeding website. During main hemorrhage, hemodilution after blood loss and subsequent volume substitute results in lowered fibrinogen ranges impairing fibrin polymerization and reduces clot stability. Thus, fibrinogen supplementation to revive plasma fibrinogen is key to normalizing clotting operate. In this case, cryoprecipitate or fibrinogen concentrates are a better possibility to revive enough plasma levels (200 mg/dL) and have to be considered when treating life-threatening bleeding. Fibrinogen could be repleted by cryoprecipitate; 1 unit per 10 kg increases fibrinogen by 50 to 70 mg/dL. Recombinant Coagulation Products Recombinant proteins are becoming extra readily available for managing bleeding, topical hemostasis and for other therapeutic interventions. The advanced function that transfusion therapy has in producing adverse outcomes is rising in the scientific literature. In 72% of the 50 reported deaths, thromboembolism was considered the probable trigger. The major endpoints were the number of sufferers suffering critical severe adverse occasions. Collagen sponges can be found in different commercial types and are derived from bovine Achilles tendon or bovine pores and skin. Bovine thrombin at present should be prevented because of its potential for antibovine thrombin antibody formation and immune-mediated coagulopathy. Fibrin sealants, also known as biologic glue or fibrin tissue adhesives, are component merchandise that combine thrombin (mostly human) and fibrinogen (usually plasma derived). They are packaged with a dual-syringe delivery system that mixes the parts to form a fibrin clot. The rising use of anticoagulation brokers creates a need for a number of pharmacologic approaches as reviewed in the chapter on anticoagulation. Topical Hemostatic Agents Topical hemostatic agents are used intraoperatively to advertise hemostasis at the web site of vascular harm and are classified primarily based on their mechanism of action. They embrace bodily and mechanical agents, caustic agents, biologic bodily brokers, and physiologic agents. Full-dose aprotinin use in coronary artery bypass graft surgery: an evaluation of perioperative pharmacotherapy and affected person outcomes. Protamine reversal of heparin affects platelet aggregation and activated clotting time after cardiopulmonary bypass. The impression of heparin concentration and activated clotting time monitoring on blood conservation.
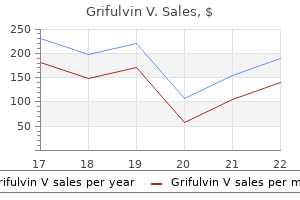
Buy genuine grifulvin v online. Fungal infection Ayurvedic medicine and chemical product antifungal cream.
When administration is discontinued, the drug is cleared from plasma in about 36 hours. Glipizide Glipizide stimulates insulin secretion over a 12-hour interval after a morning oral dose. Unlike glyburide, metabolism of glipizide within the liver produces inactive substances which would possibly be excreted in urine. Relatively speedy Table 38-5 Comparison of Sulfonylurea Therapy with Insulin Therapy Sulfonylurea Failed preliminary response in 10% to 15% of sufferers Secondary failure rate each year among handled sufferers is about 10% Hypoglycemia could additionally be more extreme Associated cardiac complications Patients may favor oral medicine Insulin No most dose Hypoglycemia could also be more frequent Lipid ranges lowered Patients could resist injections Chapter 38 � Drugs that Alter Glucose Regulation 755 clearance from the plasma minimizes the potential for long-lasting hypoglycemia. Glimepiride Glimepiride decreases blood glucose concentrations by stimulating launch of insulin from the pancreas and may decrease hepatic glucose manufacturing. Tolbutamide Tolbutamide is the shortest performing and least potent sulfonylurea (see Table 38-4). Acetohexamide Acetohexamide differs from different sulfonylureas in that the majority of its hypoglycemic action comes from its principal metabolite hydroxyhexamide, which is 2. After oral ingestion, peak plasma concentrations of acetohexamide and its energetic metabolite happen after 1. Acetohexamide is the only sulfonylurea with uricosuric properties (urocosuric medicine improve the excretion of uric acid in the urine), making it an applicable drug for the diabetic affected person with gout. Chlorpropamide Chlorpropamide is the longest appearing sulfonylurea, with a length of motion that will strategy 72 h ours (see Table 38-4). Risk factors for the development of hyponatremia include age older than 60 years, feminine gender, and the concomitant administration of thiazide diuretics. If all these danger factors are current, the frequency of hyponatremia will increase threefold. Repaglinide and nateglinide should be administered 15 to 30 minutes earlier than a meal and may never be ingested whereas fasting. Flatulence, belly cramping, and diarrhea are side effects that incessantly end result from undigested carbohydrates that reach micro organism within the decrease colon. With the exception of occasional will increase in liver transaminases, these medication are thought-about unhazardous. The accumulation of extracellular fluid as edema is undesirable in patients with congestive coronary heart failure. Although rosiglitazone has been related to cardiovascular risk, notably heart failure, this threat could also be similar to the cardiovascular dangers observed with different standard diabetes medicines. Report of the professional committee on the prognosis and classification of diabetes mellitus. Less hypoglycemia with insulin glargine in intensive insulin remedy for sort 1 diabetes. The 2 defects of kind 2 diabetes: combining medicine to deal with each insulin deficiency and insulin resistance. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on sort 2 diabetes mellitus: an algorithm for glycemic control. Lactic acidosis as a severe perioperative complication of antidiabetic biguanide treatment with metformin. Dose-response relation between sulfonylurea medication and mortality in type 2 diabetes mellitus: a p opulation-based cohort study. Other Medications Colesevelam (bile acid sequestrant) and bromocriptine mesylate (dopamine receptor agonist) lower glucose ranges and reduce HbA1c values, however the mechanisms are unclear. Neither of these medications is associated with hypoglycemia and both might cause gastrointestinal intolerance. For instance, insulin resistance in the liver is decreased with metformin whereas insulin secretion is elevated with sulfonylureas or meglitinide. The primary goal of mixture therapy is to lower HbA1c; reductions within the day by day insulin dose are a secondary benefit. Chapter 38 � Drugs that Alter Glucose Regulation diabetic patients with acute myocardial infarction. Mortality and cardiovascular threat associated with completely different insulin secretagogues in contrast with metformin in type 2 diabetes, with or and not utilizing a earlier myocardial infarction: a nationwide examine. Glyburide: second-generation sulfonylurea hypoglycemic agent; history, chemistry, metabolism, pharmacoki- 757 netics, scientific use and antagonistic effects. Moitra Hypothyroidism the first remedy of hypothyroidism is hormone substitute remedy. Therefore, measuring free T3 and T4 is important to evaluate the efficacy of treatment. A large number of substances interfere with the synthesis of thyroid hormones or scale back the quantity of thyroid tissue. These compounds include (a) thionamides, (b) inhibitors of the iodide transport mechanism, (c) iodide, and (d) radioactive iodine. Synthetic Thyroxine (T4: Levothyroxine) Synthetic thyroxine (T4) is the remedy of alternative for primary hypothyroidism. In the peripheral tissues, T4 is deiodinated to form triiodothyronine (T3; the energetic form of thyroid hormone). Although formulations of T4 (Synthroid, Levoxyl, generic preparations) may have minor variations in bioavailability, one research means that bioequivalence among formulations could additionally be equal. Thionamides (Methimazole, Propylthiouracil, Carbimazole) Thi namides are antithyroid drugs that inhibit the formation of thyroid hormone by inhibiting thyroid peroxidase to stop incorporation of iodine into tyrosine residues of thyroglobulin. In addition to blocking hormone synthesis, propylthiouracil also inhibits the peripheral deiodination of T4 and T3. The half-life of methimazole (4 to 6 hours, dosed once daily) is longer than the half-life of propylthiouracil (75 m inutes, dosed a quantity of times per day). Drug-induced decreases in excessive thyroid activity normally require a number of days, as a end result of preformed hormone T3 Formulations (Liothyronine) Liothyronine is the levorotatory isomer of T3 and is 2. In a number of patients, especially these with extreme hyperthyroidism, definite enchancment is obvious in 1 to 2 days. Side Effects Minor unwanted effects of thionamide remedy are observed in approximately 5% of sufferers and include urticarial or macular skin rash, arthralgias, and gastrointestinal discomfort. Fever or pharyngitis will be the earliest manifestation of the development of agranulocytosis. Hepatic toxicity has been reported with thionamide use, significantly propylthiouracil. Placental passage, nonetheless, is limited for propylthiouracil, making it the preferred drug to be used in the parturient. Indeed, the most important clinical effect of excessive doses of iodide is inhibition of the release of thyroid hormone. Indeed, the mixture of oral potassium iodide and propranolol is a recommended strategy. This isotope is quickly and effectively trapped by thyroid gland cells, and the next emission of damaging b rays acts almost exclusively on these cells, with little or no injury to surrounding tissue. For this purpose, iatrogenic hypothyroidism have to be thought-about preoperatively in any affected person who has previously been handled with 131I. The use of 131I is contraindicated during being pregnant as a end result of the fetal thyroid gland would focus the isotope. As a outcome, the therapeutic effectiveness of 131I for therapy of thyroid most cancers is restricted. American Thyroid Association, Endocrine Society, American Association of Clinical Endocrinologists. The effect of iodide on serum thyroid hormone levels in regular individuals, in hyperthyroid sufferers, and in hypothyroid sufferers on thyroxine replacement. Fifty years of experience with propylthiouracil-associated hepatotoxicity: what have we discovered Moitra Preparations that include artificial hormones identical to those secreted endogenously by endocrine glands could also be administered as medicine. These synthetic hormones resemble the endogenous substances in structure and activity. Corticosteroids the actions of corticosteroids are classifi d based on the potencies of these compounds to (a) evoke distal renal tubular reabsorption of sodium in exchange for potassium ions (mineralocorticoid effect) or (b) produce an antiinflammatory response (glucocorticoid effect). Several artificial corticosteroids can be found, principally for use to supply antiinflammatory effects. Modifications of structure, corresponding to introduction of a double bond in prednisolone and prednisone, have resulted in synthetic corticosteroids with stronger glucocorticoid effects than the 2 intently associated pure hormones, cortisol and cortisone, respectively (Table 40-1). Despite increased antiinflammatory effects, it has not been possible to separate this response from alterations in carbohydrate and protein metabolism. This means that the a quantity of manifestations of drug-induced glucocorticoid effects are mediated by the identical receptor. Mineralocorticoid receptors are current in distal renal tubules, colon, salivary glands, and the hippocampus.